来源:博雅
日期: 2018.12.26
访问量: 0
迄今为止,美国和欧盟已经相继批准了两种CAR-T细胞疗法,分别是诺华与宾夕法尼亚大学合作开发的Kymriah™(CTL019),用于治疗急性淋巴细胞白血病(ALL),以及 吉利德旗下公司Kite制药开发的Yescarta™,用于治疗治疗复发或难治性弥漫性大B细胞淋巴瘤(DLBCL)和原发性纵隔大B细胞淋巴瘤(PMBCL)成人患者。
尽管CAR-T的安全性和有效性已得到很好的验证,然而,由于产品的异质性、制造复杂和缺乏标准化的全球监管,CAR-T仍然是局限于数百名患者的精品疗法。
实现大规模CAR-T生产是提高其可及性的重要课题,为此,标准化各国的CAR-T制造和法规势在必行且具有挑战性。
近日,江南大学的戴晓峰教授和解放军总医院的韩为东教授作为共同通讯作者在Biotechnology Advances(SCI一区,IF=11.452)发表了一篇题为:“Standardizing CAR-T therapy: Getting it scaled up(标准化CAR-T疗法:扩大规模)”的文章,围绕限制CAR-T的生产规模的问题,回顾了当下的状况和挑战。

标准化CAR-T制造
基因工程T细胞的临床制备首先需要采集患者外周血单个核细胞,对其进行增殖,然后进行基因修饰和扩增,并在输注前进行筛选和/或冷冻保存。鉴于CAR-T生产原料的多样性和现有的各种培养技术,修饰T细胞终的质量变化仍然是一个有待解决的重要问题。
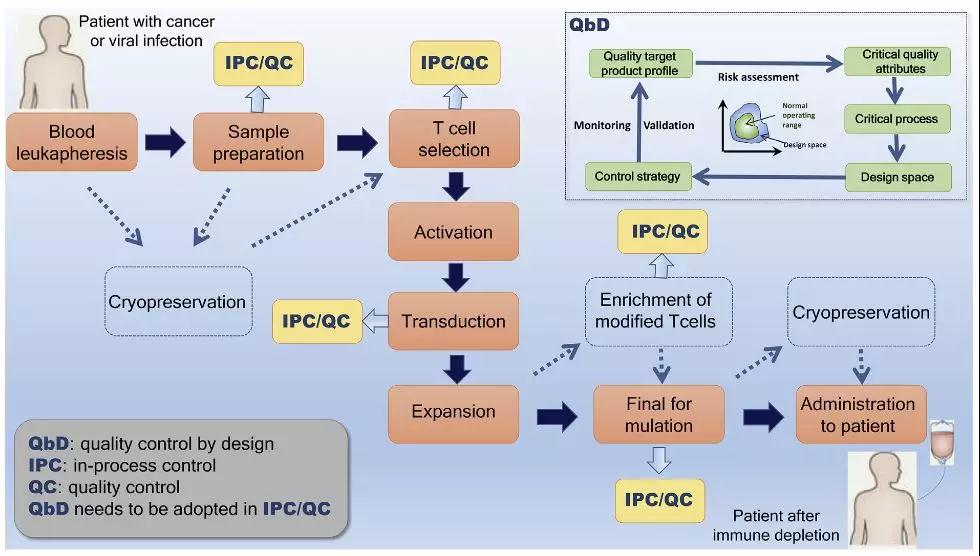
CAR-T细胞生产的经典工作流程(图片来源:Biotechnology Advances)
戴晓峰教授和韩为东教授指出,以下7个因素的标准化对于大规模CAR-T生产的成功是决定性的:
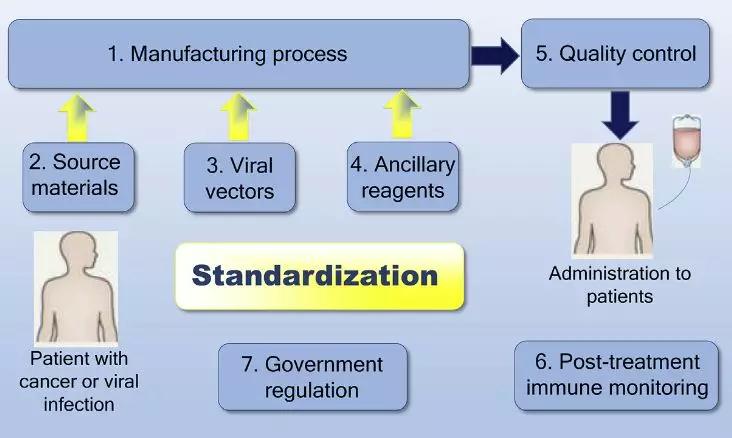
7个决定性因素(图片来源:Biotechnology Advances)
制造过程
制造过程应该追求(1)生产安全且临床上有效的细胞产品;(2)可扩展性,以便更多的患者可以从这些技术中受益而不牺牲产品的质量和可再生产性。
为了实现这一目标,文中倡议使用封闭式自动化系统和质量源于设计(quality-by-design,QbD)理念。
封闭式自动化系统
实施封闭的培养系统是一个防止产品污染和保持无菌环境的可靠策略。为此,可以将关键试剂的生产程序纳入一个封闭系统,或者,建立良好的跟踪供应链和技术,使原料生产能够在封闭系统中完成。
从细胞制备、筛选、培养、扩展、转导到配方,自动化在标准化生产过程中有着特殊的用途,目前已经有一些公司建立相应的技术平台。
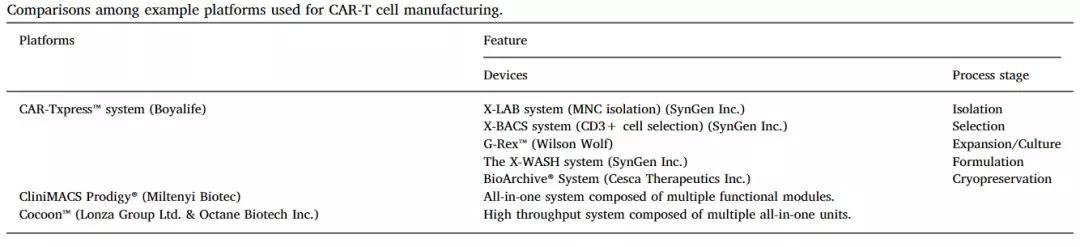
几种CAR-T制造平台的对比
(图片来源:Biotechnology Advances)
- 中国生物技术公司博雅控股(BoyaLife)建立一个集临床级细胞分离、纯化、培养、洗涤和配方于一体的CAR-TXpress™平台,这是一种紧凑的单元操作装置。
- 德国美天旎生物(Miltenyi Biotec)自主研发的CliniMACS® Prodigy是一个集细胞分选、扩增、转导、培养等多功能于一身的高度自动化临床级别细胞制备平台,提供一种由多个功能模块组成的一体化系统。
但其使用受到一些缺点的限制,例如成本高,长时间细胞培养期间其他功能部分不可用,以及需要定制供应商设计软件。
- 瑞士CDMO巨头Lonza与Octane Biotech合作开发自动生物反应器Co-coon™,这是一个由多个一体化单元组成的高通量系统,可以通过可定制规模的高通量方式并行处理源细胞,被称为GMP-in-a-box系统。
QbD
QbD始于所需的产品质量属性的描述,这直接确定了影响产品安全性和有效性的特征和相关参数,并为量化参数变异创造了设计空间。通过将科学知识和风险分析整合到制造过程的开发中,QbD可识别合适的操作信封(envelope),并且可通过知识的增加进行迭代修改,从而生产高质量的产品。
鉴于CAR-T产品的异质性和产品质量的不确定性,如何将细胞群的可测量分子和细胞特征与产品质量联系起来,将决定CAR-T制造与QbD的成功与否。
原料
CAR-T的原料是高度可变的患者细胞,通常来自接受放化疗的患者,这决定了细胞中可能包含不同水平的抑制因子或对刺激具有抗性的T细胞亚群。这些因素对确定制造过程的可重复性造成挑战。
目前,两种策略可以解决这一挑战。首先,从T细胞中除去抑制元件。已经报道了通过几种方法去除这些抑制元件后T细胞培养物的改善结果,例如:(1)通过同时使用两种细胞表面标志物CD44和CD137分离抗肿瘤CD4 T细胞,耗尽抑制性调节性T细胞群(Treg细胞);(2)使用涂有抗CD3和抗CD28的大磁珠与适用于袋子的大磁铁,这代表了一种将隔离和刺激结合在一起的强大的制造工艺。
其次,起始原料使用定义的T细胞子集。特定的T细胞亚群,即记忆干细胞,被证明可以提供改善的临床效果并被认为是良好的起始原料。例如,针对巨细胞病毒或Epstein-Barr病毒的内源性抗原反应性记忆T细胞已被用作基因工程的来源T细胞,因为具有抗原特异性的记忆T细胞有利于预防病原体,因为它们在淋巴细胞清除和CAR-T后防止病毒再激活方面具有良好的效果。
病毒载体
病毒载体用于将CAR转导入T细胞的过程,被认为是美国CAR-T细胞制造过程的关键原料。与高度异质和个性化的血液样本和CAR-T细胞终产品不同,病毒载体可以大量生产并在-80°C下稳定储存4至9年。
通常,批量生产用于细胞疗法的病毒载体的时间超过2周,其中大部分时间花费在产生足够量的细胞上。细胞培养扩增数天至适当的数量,然后进行质粒转染,生产病毒载体。病毒载体应该具有阻止它们重新获得复制能力的特征。密码子优化的Gag/Pol可大限度地减少载体组分之间的同源性,自我灭活长末端重复序列,去除所有不必要的序列和辅助基因,这些都是用于预防这一问题的规范设计。
转染后48小时内,表达CAR的病毒载体可从培养基中收获,通过交换培养基可以收获多批病毒载体。在过滤生产细胞和碎片后,病毒载体通过一系列下游处理进行纯化,并使用储存缓冲液进行富集。
载体质量包括无菌和纯度(即缺乏包装细胞)、效力、同一性和滴度,对于终CAR-T产物的成功至关重要,为此,需要在受控条件下、在GMP设施中以小的开放处理制造,以及随后的一系列安全性测试以确保每批载体产品符合用于T细胞转导之前的标准。
根据FDA指南,安全性测试涉及“细胞特性测试”,其通过一系列表型和/或生物化学检测来评估细胞特性和异质性。“效能测试”评估CAR-T产品的功能;“活力测试”定量细胞活力;“无菌测试”验证细胞没有被外来物质污染,如细菌、真菌、支原体和病毒;“纯度测试”确保产品不含残留的宿主细胞和质粒DNA,并免除与工艺相关的杂质; “一般安全性测试”评估终产品的临床应用的安全性,以及随后在给药前应用于解冻冷冻细胞的“批次释放测试”。
当细胞转导中使用整合病毒载体时,需要进行检查细胞和培养基中复制型逆转录病毒/慢病毒(RCRs/RCLs)是否存在的额外测试,以帮助确定终CAR-T产品对患者输液的安全性。细胞转导的载体选择和优化对于降低大规模CAR-T制造的终产品的可变性和大化效率至关重要。常用载体是基于γ逆转录病毒和慢病毒的复制缺陷型载体系统。慢病毒载体保留了感染非分裂细胞的能力,因此具有增加的转导效率。第三代微小慢病毒载体由于其体积大大减小(由一组HIV基因构建)和安全性增强(在载体生产细胞之外无功能)而获得进一步青睐。
当使用病毒载体时,应监测生产、收集和纯化步骤,并提供有关提供载体的制造商的详细信息。所有整合载体的一个潜在问题是通过将载体DNA整合到宿主细胞癌基因附近引起的插入诱变。在这方面,慢病毒载体优于γ逆转录病毒载体,因为它们的整合模式有利于远离基因启动子的位点。其他因素如编码转基因、启动子、细胞类型和适应症也与插入诱变相关。
如果载体产物中存在RCR/RCL,则可以增加致癌潜力。因此,在整个载体制造过程和载体修饰细胞产物的多个阶段,需要进行RCR/RCL的严格测试。
FDA建议对使用病毒载体的细胞和基因治疗进行长期安全性评估。例如,一项美国试验已经在进行,对接受CTL019治疗的患者进行15年随访(NCT02445222),其主要目标是监测与使用病毒载体相关的延迟不良事件。
非病毒载体
基于mRNA、DNA和转座子/转座酶的电穿孔系统已经显示出能有效转导T细胞的前景,这解决了有关病毒载体使用的一些问题,例如安全性问题、大规模制造和复杂的操作步骤。
当使用非病毒方法进行转导时,应严格控制mRNA或DNA质粒的质量,试剂应为临床级,并且电穿孔装置应适用于临床。
然而,非病毒转座子系统可能需要长时间离体扩增以使CAR+ T细胞达到治疗剂量,这在很大程度上限制了它们的临床应用。目前,已经提出了几个解决这个问题的方法。例如,非病毒睡美人(SB)转座的CAR基因来自极小化DNA载体(即小环)而非传统SB质粒可以显著提高稳定的基因转移率和细胞活力,以满足临床CAR+ T细胞对产量的要求。
辅助试剂
在离体操作过程中所使用的试剂,例如抗体、细胞因子、血清或固体支持物如珠子,其质量可能显著影响CAR-T终产品的安全性、纯度和效力。
所有试剂必须遵循USP关于辅助材料的建议。特别重要的是,鉴于制造过程的高度可变性,应限制使用天然来源的物质。例如,培养基应该是无血清的,以降低被牛海绵状脑病(BSE)或病毒污染的风险,如果不可能的话,应该提供证实血清必要性的文件。同样,推荐使用其他添加剂,如细胞因子和抗生素,以便去除。如果都不适用,那么它们应该被精确地化学定义,具有临床等级,并且没有内毒素和偶然污染。
这些添加的组件需要有效且稳定地协同工作以形成集成的试剂系统,理想的情况下,该制备系统使得制造过程足够稳健,以生产患者来源的合格细胞产品。
质量控制
CAR-T产品的安全性、纯度、特性和效力构成了其关键质量属性。鉴于其高度异质性和复杂的制造程序,个体化疗法对这些属性的要求远远高于普通的医药产品。因此,CAR-T产品必须接受许多质量控制分析,以满足所有发布的标准和良好生产规范(GMP)指南。
转导效率
转导效率(CAR阳性T细胞的百分比)是决定CAR-T细胞体内效应的重要参数。目前的指南要求通过流式细胞仪或等效途径确定转导效率,但没有明确规定该参数的阈值。
理论上,通过将抗生素抗性基因掺入CAR载体,然后使用相应的抗生素选择阳性细胞,来生产纯CAR阳性T细胞是可行的。然而,由于其对CAR-T产品施加的复杂安全问题,不适用于临床应用。
因此,仍然未确定细胞产品中是否存在佳输注的CAR-T细胞群,这需要科学界发现和制定可行的标准,以及政府当局发布控制转导效率的相应指南。
生物学效力
CAR-T细胞的生物学效力需要在输注前确定,常用指标是CAR-T细胞的裂解活性。免疫检查点,如PD-1和CTLA-4,已经表明能抑制CAR-T细胞的效力,但未包含在指南中已确定的效力控制阈值。
由于离体和体内CAR-T效力之间存在差异,并且CAR-T细胞进行的修饰增加了释放测试的复杂性,因此有必要制定标准方案来全面评估CAR-T细胞的体外生物学功能:确定改变CAR-T细胞生物学效力的分子的表达水平,并且建立公认的通用技术来控制CAR-T细胞的生物学效力。
毒性
毒性是CAR-T产品临床应用的重要考虑因素。研究人员应提供全面而详细的毒性评估标准,包括毒性等级分类('on-target','on-target, off tumor','off-target')和为每个等级制定精心设计的策略。
除了详细说明质量控制中的毒性外,在进行CAR-T治疗时,还应考虑患者护理,如制备免疫抑制药物以预防细胞因子释放综合征的发生和其他严重的不良反应。
与传统医疗产品相比,CAR-T细胞的质控过程在可用材料的数量和测试的时间点选择方面有更多限制。由于这些产品在输注或冷冻保存之前的保质期有限,因此还必须及时进行测试。
自动化
自动化有助于标准化质量控制方案(protocol):
自动染色、采集和分析过程将有益于培养细胞群的流式细胞仪分析,例如使用MACSQuant分析仪和分析工具;
自动获取和分析流量数据可以帮助快速生成标准化报告并显著减少工作量,例如LIMS(实验室信息和管理系统);
实施自动跟踪和批量记录,可以促进质量控制,其中有效的跟踪需要涵盖整个制造过程,即从开始材料采样到患者输注。在制程(in-process)和质量控制系统的紧密连接中,建立自动批量记录是必要的,这允许在每个制造过程结束时推广高度标准化的个性化方案,从而比较不同生产地点和不同流程的质量控制数据,促进我们对细胞产品的理解。
然而,尽管目前有许多选择可以实现CAR-T(“制造过程”)的某些步骤的自动化,但是使整个过程自动化的设备相对较少。因此,为了创造小化人为干预和易于使用的封闭环境,开发覆盖整个生产过程的制造系统是必要且具有挑战性的。此外,关于细胞样品自动分析的监管协议能使自动化制程中的决策成为可能,从而实现细胞治疗产品的自动化质量控制,但目前仍然缺乏。
总之,质量控制对CAR-T疗法的成功至关重要,需要建立一个由公认的指导方针监管的标准化质量控制框架,并且开发尽可能少的外部干预的自动化设备,以帮助实现这一目标。
治疗后免疫监测
与传统的现成药物不同,稳健监测过继转移后的CAR-T细胞对于其生物活性的阐明和改进至关重要。目前的监测重点是评估CAR-T细胞频率和表型,内源性免疫反应的变化,炎症细胞因子水平,肿瘤抗原表达和微环境。
CAR-T的大规模应用和患者随访涉及高通量和多重分析技术,包括TCR测序,基因表达平台,多重质谱细胞计数方法和多通道微珠免疫测定。肿瘤PET成像等新兴方法尚未应用于CAR-T细胞反应的评估,但或许能通过追踪免疫细胞为CAR-T治疗后患者反应的理解增添新的内容。
在全球范围内,标准化和协调监测分析可以使结果在不同地点的机构、公司和医院之间具有可比性,从而产生大量免疫监测信息,有助于推动CAR-T领域的进一步发展。
政府监管
迄今为止,全球已经注册了近300项CAR-T临床试验(不包括试验终止或撤销),美国有107项试验,中国有130项(根据www.clinicaltrials.gov)。鉴于独特的功能和来源,任何传统的'off-the-shelf(现成)'药物都无法代表CAR-T产品。无论是基因还是细胞疗法,都必须遵循它们的任何指导方针和行政管理规范,特别是发布的关于CAR-T细胞文件。
美国
在美国,FDA将CAR-T细胞分类为351种生物产品,并通过“Considerations for the design of early-phase clinical trials of cellular and gene therapy products”和“Guidance for industry: preclinical assessment of investigational cellular and gene therapy products”对其进行监管。
此外,CAR-T产品应遵循指南“Guidance for FDA reviewers and sponsors: content and review of chemistry, manufacturing and control (CMC) information for human somatic cell therapy investigational new drug applications (INDs)”和“Guidance for FDA reviewers and sponsors: content and review of chemistry, manufacturing and control (CMC) information for human gene therapy investigational new drug appli- cations (INDs)”,其中详细说明CAR-T细胞的制造、过程控制、释放测试和质量控制。
为了加速医疗产品开发并向需要它们的患者提供更有效的治疗,美国政府于2016年12月发布了“21世纪治愈法案(21st Century Cures Act)”,以简化药物和设备审批流程,并更快地提供临床治疗,美国政府已在9年内授权5亿美元用于帮助FDA实施该法律。在此基础上,建立了新的加速产品开发计划,包括“再生医学高级治疗(RMAT)”和“突破性医疗器械项目”,该计划为符合涵盖范围的生物制品或设备(包括CAR-T和CAR-T相关设备)提供了新的加急选择。
欧洲
在欧洲,医疗管理由欧洲药品管理局(EMA)监管,适用于CAR-T产品监管的指南是2012年终确定的“Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells”,它定义了科学原理,为开发和评估含有经授权的人类使用的基因修饰细胞的医药产品提供指导,重点放在细胞产品的质量、安全性和有效性上。此外,CAR-T细胞的制造应遵循先进治疗药品(第1394/2007号)的规定以及基因转移医药产品的质量、临床前和临床方面的指导(CPMP/BWP/3088/99)。
与FDA发布的指导原则不同,EMA有专门的文件来规范作为医疗产品的基因修饰细胞。
中国
标准化和自动化制造代表了具有更高安全性、更便宜的产品,因此在发展中国家实施CAR-T产品的规范性规定尤其有利且具有实际意义,因为它们通常较少设有自动化的“个体化车间”。
在我国,有助于规范CAR-T产品临床研究的个指南是国家卫计委于1993年发布的《人的体细胞治疗及基因治疗临床研究质控要点》。
后来,国家食品药品监督管理总局(NMPA)发布了一系列基因和细胞疗法的监管准则。2003年,NMPA发布了《人基因治疗研究和制剂质量控制技术指导原则》,其中定义了临床试验中考虑的医疗产品的基本要求,并将遗传修饰的细胞和携带功能基因的载体指定为医疗产品。NMPA在同一年发布的另一条适用于CAR-T产品的指南是《人体细胞治疗研究和制剂质量控制技术指导原则》。
此外,CAR-T产品还受到应用于临床试验的药物通用指南的监管,即由NMPA于2003年8月发布的《临床试验用药物生产质量管理规范》。监管干细胞临床试验的指南 ,即“限制临床应用医疗技术”,由国家卫计委于2015年发布。NMPA于2017年12月22日发布了《细胞治疗产品研究与评价指导原则》(试行,第216号,2017年)。
尽管已经发布不少法规,但为了安全性的考虑,CAR-T细胞还有一些独特的特征需要受到监管,而目前我国的任何监管指南都没有涵盖这些特征,如转导效率,预计CAR-T细胞的行政文件规范将在不久的将来发布。
为整个CAR-T行业制定广为接受的指南具有挑战性,因为具有不同表型或生产方法的CAR-T细胞通常会产生相同的效果。因此,良好的指导方针应保证CAR-T产品的安全性和有效性,使其制造透明化和标准化,并描述生产、制程释放测试和认证分析的低和基本标准。
另一方面,一些决定CAR-T产品的效率和安全性的因素,如果没有作为政府指导原则发布,则需要作为建议发布,以帮助保证和改善CAR-T产品的质量。例如,预处理,通常涉及去除抑制抗肿瘤活性的各种抑制因子的非清髓性化学治疗方案,这应该需要考虑在未来的指南更新中加入。其他因素,如修饰T细胞的活力、表型、无菌、同质性和遗传稳定性,以及每个细胞的基因拷贝数也可以在指南作出建议。
目前,部分国家已发布了CAR-T细胞临床试验的国内指南,并且不同国家的监管部门之间进行交换是很常见的,例如在FDA和EMA之间以及FDA和CFDA之间的。
为了实现全球范围的临床试验,各国的监管指南需要实现协调。全球药品监管机构的九名成员,包括FDA、EMA、加拿大卫生部、巴西ANVISA、印度国家生物制品研究所(INB)、日本卫生福利和劳工/制药和医疗器械局、韩国食品药品安全部、新加坡卫生科学局(HSA)和Swissmedic已召集组建论坛,讨论细胞或基因治疗监管的佳监管方案(https://www.i-p-r-f.org/en/working-groups/gene- therapy-workinggroup/)。在制定统一的法规之前,CAR-T产品开发中依旧存在很大的不确定性,这在很大程度上取决于监管机构的主观判断。
相关新闻
前沿动态
行业政策

在线咨询
留言您的联系方式及需求,专业顾问将于收到资料后尽快与您回复。
我要预约
填写您的预约需求,您将获得相应的专业顾问满足您的需求。
电话咨询
全国统一客服专线400-885-0012,期待为您服务。
微信咨询
直接添加客服微信号,专业顾问随时准备解答您的一切疑问。
在线咨询


服务热线
400-885-0012


官方微信


TOP

观望只会错过守护宝宝和家庭的机会
存储新生儿干细胞
机会一生只有一次

扫码了解细胞新时代